![]()
CpG_Me is a WGBS alignment pipeline for a SLURM job scheduler on a high-performance computing cluster. CpG_Me takes raw fastq files and generates CpG methylation count matrices (Bismark genome-wide cytosine reports). Scripts are available for both paired end (PE) and single end (SE) sequencing approaches. The extracted CpG methylation count matrices can be used for the identification of differentially methylated regions (DMRs) through the accompanying DMRichR workflow.
Table of Contents
- Overview
- Quick Start
- Installation
- Merging Lanes
- Correcting for Methylation Bias (m-bias)
- Run the Pipeline
- QC Report
- DMR Calling
- Citation
- Publications
- Acknowledgements
Overview
A single command line call runs the main pipeline for all samples and a final call generates html QC/QA reports for all samples.
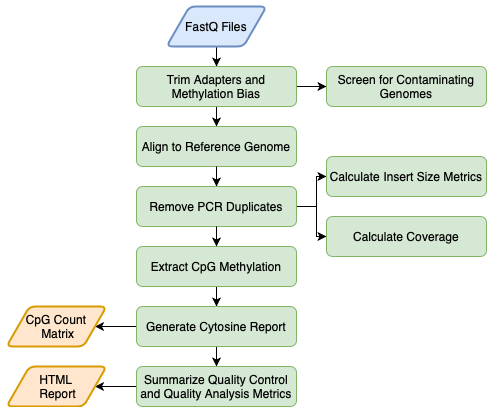
Quick Start
Assuming everything is installed and that you used have 150 bp paired-end reads from the NovaSeq, all you need to do is change to your working directory and run the following command for the human genome:
sbatch --array=1-96 /share/lasallelab/programs/CpG_Me/Paired-end/CpG_Me_PE_controller.sh hg38
Once your alignments are complete, you can generate the overall QC/QA report with the following command:
sbatch /share/lasallelab/programs/CpG_Me/Paired-end/CpG_Me_PE_QC.sh
Installation
This workflow utilizes the following packages, which need to be installed and in your path:
Bioconda can be used to install the needed programs:
conda install -c bioconda trim-galore bismark bowtie2 fastq-screen picard multiqc
Bisulfite converted genomes will also have to be created and placed in an external folder for the genome of interest as well as the genomes you would like to use to screen for contamination. This can be accomplished by using bismark_genome_preperation, which is detailed in the Bismark docs, and example scripts are available in the Genome_preperation folder of this repository. These scripts expect that each bisulfite converted genome is located in a genomes folder, which contains a folder for each genome within it (i.e. hg38). However, you can also download the prepared indices for a number of genomes via FastQ Screen with the command fastq_screen --bisulfite --get_genomes.
The genome folder structure should appear as:
├── genomes
│ ├── hg38
│ │ ├── Bisulfite_Genome
│ │ │ ├── CT_conversion
│ │ │ ├── GA_conversion
│ │ ├── Bowtie2
│ │ ├── hg38.fa
│ │ ├── hg38.fa.fai
│ ├── mm10
│ │ ├── Bisulfite_Genome
│ │ │ ├── CT_conversion
│ │ │ ├── GA_conversion
│ │ ├── Bowtie2
│ │ ├── mm10.fa
│ │ ├── mm10.fa.fai
The paths will also need to be changed in the controller, switch, and QC scripts through the mainPath variable, where for our environment they begin with /share/lasallelab/ and the scripts themselves are located in the programs folder. Finally, the paths will also need to be changed in fastq_screen.conf and multiqc_config.yaml configuration files.
The overall folder structure should appear as:
├── programs
│ ├── CpG_Me
│ │ ├── Genome-preperation
│ │ ├── Paired-end
│ │ ├── Single-end
│ │ ├── fastq_screen.conf
│ │ ├── README.md
│ │ ├── LICENSE
├── genomes
│ ├── hg38
│ ├── mm10
Merging Lanes
The most straightforward approach to merging the results of multiple lanes of data for the same sample is as follows (see ref and FASTQ_Me):
-
Check for the right number of unique sample IDs for both R1 and R2
countFASTQ(){ awk -F '_' '{print $1}' | \ sort -u | \ wc -l } export -f countFASTQ R1=`ls -1 *R1*.gz | countFASTQ` R2=`ls -1 *R2*.gz | countFASTQ` if [ ${R1} = ${R2} ] then lanes=`ls -1 *R1*.gz | \ awk -F '_' '{print $1}' | \ sort | \ uniq -c | \ awk -F ' ' '{print $1}' | \ sort -u` echo "${R1} samples sequenced across ${lanes} lanes identified for merging" else echo "ERROR: There are ${R1} R1 files and ${R2} R2 files" exit 1 fi -
Create file of unique IDs based on _ delimiter and first string (use file for CpG_Me)
ls -1 *fastq.gz | \ awk -F '_' '{print $1}' | \ sort -u > \ task_samples.txt -
Test merge commands for each read (look over each one carefully)
mergeLanesTest(){ i=$1 echo cat ${i}\_*_R1_001.fastq.gz \> ${i}\_1.fq.gz echo cat ${i}\_*_R2_001.fastq.gz \> ${i}\_2.fq.gz } export -f mergeLanesTest cat task_samples.txt | parallel --jobs 6 --will-cite mergeLanesTest -
Use merge commands for each read by removing echo and the escape character on >
mergeLanes(){ i=$1 cat ${i}\_*_R1_001.fastq.gz > ${i}\_1.fq.gz cat ${i}\_*_R2_001.fastq.gz > ${i}\_2.fq.gz } export -f mergeLanes cat task_samples.txt | parallel --jobs 6 --verbose --will-cite mergeLanes
Now, not only are your samples merged across lanes, but you now also have your task_samples.txt file for the next steps. If your data is single end then you need to modify accordingly, where you will also need to slightly modify the task_samples.txt file after too.
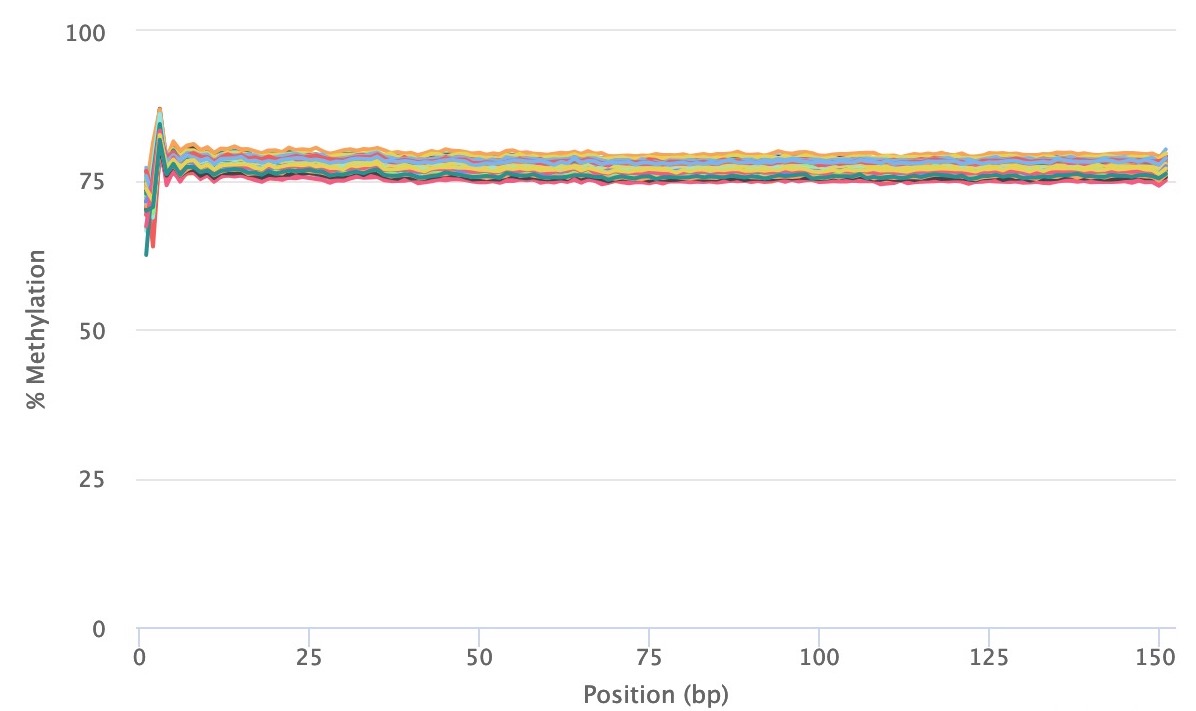
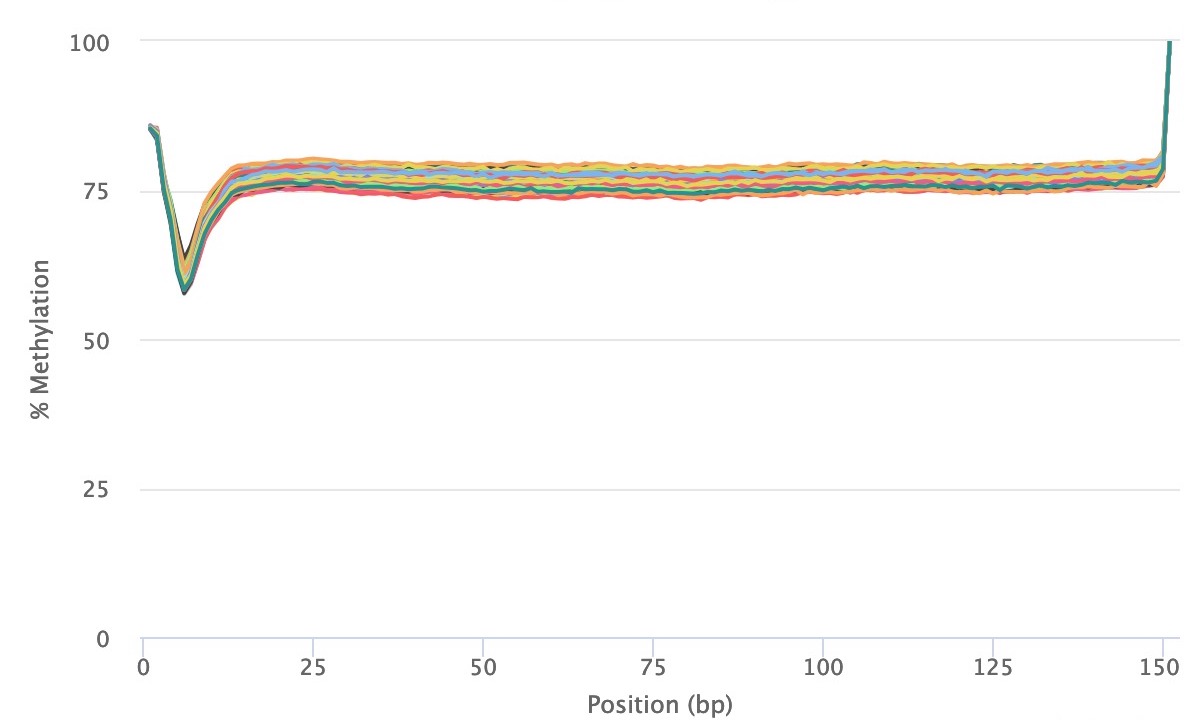
Correcting for Methylation Bias (m-bias)
Methylation bias (m-bias) is a technical artifact where the 5’ and 3’ ends of reads contain artificial methylation levels due to the library preparation method (see Figure 2 in Hansen et al.). One example is the random priming used in post-bisulfite adapter tagging (PBAT) methods (read more here). In paired-end sequencing approaches, the m-bias can also differ between reads 1 and 2 (read more here). Therefore, it is important to always examine for this bias in the MultiQC reports. CpG m-bias can be used to guide trimming options, while CpH m-bias can be used to judge for incomplete bisulfite conversion. In our experience, we have come across the following parameters, although we recommend to examine every dataset, particularly when trying a new library preparation method or sequencing platform. In paired end approaches, the 5’ end of read 2 tends to show the largest m-bias.
To address m-bias the provided scripts have reasonable defaults set, which are the first choices in the tables below. However, the CpG_Me_switch.sh script can be customized for different library preparation methods:
Paired End (PE)
| Library preparation kit | clip_r1 | clip_r2 | three_prime_clip_r1 | three_prime_clip_r2 |
|---|---|---|---|---|
| Accel-NGS Methyl-Seq Kit (Swift) | 10 | 20 | 10 | 10 |
| TruSeq DNA Methylation Kit (EpiGnome) | 8 | 20 | 8 | 8 |
Single End (SE)
| Library preparation kit | clip_r1 | three_prime_clip_r1 |
|---|---|---|
| TruSeq DNA Methylation Kit (EpiGnome) | 8 | 8 |
| MethylC-Seq (Original Method) | 7 | 10 |
M-Bias Examples
Read 1
Read 2
Run the Pipeline
- Create a parent directory for the project
-
Within that parent project directory, add a text file called “task_samples.txt”, where each new line contains the entire sample name exactly as it appears on the fastq read pair files, aside from the end part (“_1.fq.gz” or “_2.fq.gz”). Only name a sample once, not twice, and make sure it is .fq.gz and not fastq.gz. Also, if you’re using excel or a windows desktop, you will need to change the linebreaks from windows to unix, which can be done using BBedit (File > Save As… > Line Breaks > Unix) or on command line (but make sure the files have different names):
awk '{ sub("\r$", ""); print }' task_samples_windows.txt > task_samples.txt -
Within that parent directory create a folder called “raw_sequences” that contains all raw paired fastq files (sampleID_1.fq.gz and sampleID_2.fq.gz)
Overall, the directory tree structure should be the following:
├── Project │ ├── raw_sequences │ │ ├── sample1_1.fq.gz │ │ ├── sample1_2.fq.gz │ │ ├── sample2_1.fq.gz │ │ ├── sample2_2.fq.gz │ ├── task_samples.txt - Ensure the trimming options in the switch script are appropriate for the Methylation Bias (m-bias) of your library preparation method as well as your sequencing chemistry. The HiSeq and MiSeq series of sequencers use a 4 color chemistry, while NovaSeq and NextSeq series use a 2 color chemistry. For a 4 color chemistry you should use
--quality 20, while for 2 color chemistry you should use--2colour 20.
Now with that structure in place it’s ready to run, so from the parent directory, modify and run this command:
sbatch --array=1-12 /share/lasallelab/programs/CpG_Me/Paired-end/CpG_Me_PE_controller.sh hg38
Let’s break this apart:
- sbatch is how you submit a job to a HPCC with a slurm workload manager
- –array=12 lets you specify the number of samples, as well as subset. Here we are running samples 1 to 12. You could run select samples using the following format –array=2,4-12
- The next call is the location of the executable shell script that will schedule all jobs with proper resources and dependencies on a per sample basis
- Genome (hg38, rheMac8, mm10, rn6)
Single End (SE) Sequencing
For single end sequencing, follow the same approach as above but with minor changes.
The directory should appear as:
├── Project
│ ├── raw_sequences
│ │ ├── sample1.fq.gz
│ │ ├── sample2.fq.gz
│ ├── task_samples.txt
The calls to the scripts would be:
sbatch --array=1-12 /share/lasallelab/programs/CpG_Me/Single-end/CpG_Me_SE_controller.sh hg38
QC Report
There is also a final html QC report that should be run after all samples have finished, which you also need to launch from the working directory. To generate the QC report for paired end sequencing data, the command is:
sbatch /share/lasallelab/programs/CpG_Me/Paired-end/CpG_Me_PE_QC.sh
To generate the QC report for single end sequencing data, the command is:
sbatch /share/lasallelab/programs/CpG_Me/Single-end/CpG_Me_SE_QC.sh
These reports can be customized by modifying the multiqc_config.yaml files.
DMR Calling
Statistical testing for differentially methylated regions (DMRs) can be achieved by DMRichR, which utilizes the cytosine_reports folder created by CpG_Me.
Citation
If you use CpG_Me in published research please cite the 4 following articles:
Laufer BI*, Neier KE*, Valenzuela AE, Yasui DH, Lein PJ, LaSalle JM. Placenta and Fetal Brain Share a Neurodevelopmental Disorder DNA Methylation Profile in a Mouse Model of Prenatal PCB Exposure. Cell Reports, 2022. doi: 10.1016/j.celrep.2022.110442
Krueger F, Andrews SR. Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics, 2011. doi: 10.1093/bioinformatics/btr167
Matrin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal, 2011. doi: 10.14806/ej.17.1.200
Ewels P, Magnusson M, Lundin S, Käller M. MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics, 2016. doi: 10.1093/bioinformatics/btw354
Publications
The following publications utilize CpG_Me:
Laufer BI*, Neier KE*, Valenzuela AE, Yasui DH, Lein PJ, LaSalle JM. Placenta and Fetal Brain Share a Neurodevelopmental Disorder DNA Methylation Profile in a Mouse Model of Prenatal PCB Exposure. Cell Reports, 2022. doi: 10.1016/j.celrep.2022.110442
Zhu Y, Gomez JA, Laufer BI, Mordaunt CE, Mouat JS, Soto DC, Dennis MY, Benke KS, Bakulski KM, Dou J, Marathe R, Jianu JM, Williams LA, Gutierrez Fugon OJ, Walker CK, Ozonoff S, Daniels J, Grosvenor LP, Volk HE, Feinberg JI, Fallin MD, Hertz-Picciotto I, Schmidt RJ, Yasui DH, LaSalle JM. Placental Methylome Reveals a 22q13.33 Brain Regulatory Gene Locus Associated with Autism. Genome Biology, 2022. doi: 10.1186/s13059-022-02613-1
Laufer BI, Hasegawa Y, Zhang Z, Hogrefe CE, Del Rosso LA, Haapanan L, Hwang H, Bauman MD, Van de Water JA, Taha AY, Slupsky CM, Golub MS, Capitanio JP, VandeVoort CA, Walker CK, LaSalle JM. Multi-omic brain and behavioral correlates of cell-free fetal DNA methylation in macaque maternal obesity models. bioRxiv preprint. doi: 10.1101/2021.08.27.457952
Brown AP, Cai L, Laufer BI, Miller LA, LaSalle JM, Ji, H. Long-term effects of wildfire smoke exposure during early life on the nasal epigenome in rhesus macaques. Environment International, 2022. doi: 10.1016/j.envint.2021.106993
Laufer BI*, Gomez JA*, Jianu JM, LaSalle, JM. Stable DNMT3L Overexpression in SH-SY5Y Neurons Recreates a Facet of the Genome-Wide Down Syndrome DNA Methylation Signature. Epigenetics & Chromatin, 2021. doi: 10.1186/s13072-021-00387-7
Maggio, AG., Shu, HT., Laufer, BI., Hwang, H., Bi, C., Lai, Y., LaSalle, JM., Hu, VW. Impact of exposures to persistent endocrine disrupting compounds on the sperm methylome in regions associated with neurodevelopmental disorders. medRxiv preprint, 2021. doi: 10.1101/2021.02.21.21252162
Mordaunt CE, Jianu JM, Laufer BI, Zhu Y, Dunaway KW, Bakulski KM, Feinberg JI, Volk HE, Lyall K, Croen LA, Newschaffer CJ, Ozonoff S, Hertz-Picciotto I, Fallin DM, Schmidt RJ, LaSalle JM. Cord blood DNA methylome in newborns later diagnosed with autism spectrum disorder reflects early dysregulation of neurodevelopmental and X-linked genes. Genome Medicine, 2020. doi: 10.1186/s13073-020-00785-8
Laufer BI, Hwang H, Jianu JM, Mordaunt CE, Korf IF, Hertz-Picciotto I, LaSalle JM. Low-Pass Whole Genome Bisulfite Sequencing of Neonatal Dried Blood Spots Identifies a Role for RUNX1 in Down Syndrome DNA Methylation Profiles. Human Molecular Genetics, 2020. doi: 10.1093/hmg/ddaa218
Wöste M, Leitão E, Laurentino S, Horsthemke B, Rahmann S, Schröder C. wg-blimp: an end-to-end analysis pipeline for whole genome bisulfite sequencing data. BMC Bioinformatics, 2020. doi: 10.1186/s12859-020-3470-5
Lopez SJ, Laufer BI, Beitnere U, Berg E, Silverman JL, Segal DJ, LaSalle JM. Imprinting effects of UBE3A loss on synaptic gene networks and Wnt signaling pathways. Human Molecular Genetics, 2019. doi: 10.1093/hmg/ddz221
Vogel Ciernia A*, Laufer BI*, Hwang H, Dunaway KW, Mordaunt CE, Coulson RL, Yasui DH, LaSalle JM. Epigenomic convergence of genetic and immune risk factors in autism brain. Cerebral Cortex, 2019. doi: 10.1093/cercor/bhz115
Laufer BI, Hwang H, Vogel Ciernia A, Mordaunt CE, LaSalle JM. Whole genome bisulfite sequencing of Down syndrome brain reveals regional DNA hypermethylation and novel disease insights. Epigenetics, 2019. doi: 10.1080/15592294.2019.1609867
Acknowledgements
The development of this program was suppourted by a Canadian Institutes of Health Research (CIHR) postdoctoral fellowship [MFE-146824] and a CIHR Banting postdoctoral fellowship [BPF-162684]. Contributions were made by Hyeyeon Hwang and Charles Mordaunt. I would also like to thank Matt Settles and Ian Korf for invaluable discussions related to the bioinformatic approaches utilized in this repository.