Genetic Impacts on the Epigenome
 RUNX1 Profile
RUNX1 Profile
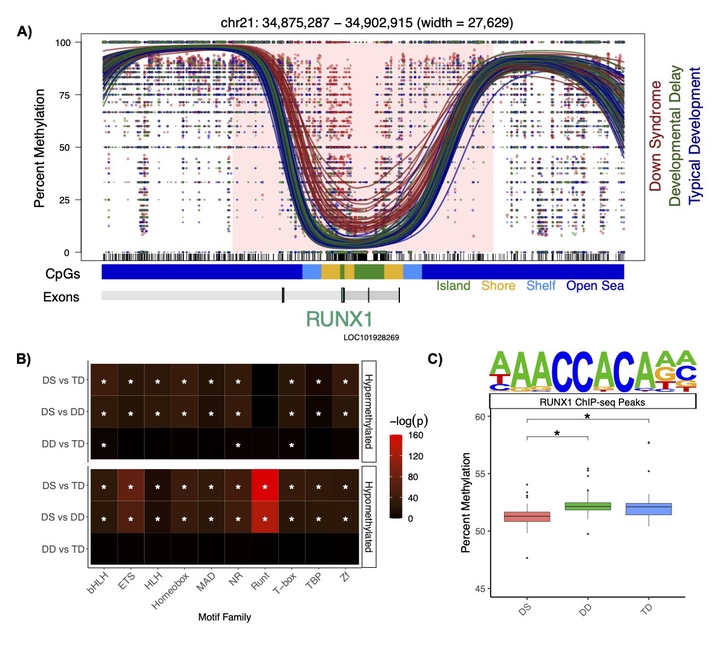
Down syndrome (DS) is the leading genetic cause of NDDs and there is variation in the level of intellectual disability amongst patients. While DS is caused by trisomy 21, it represents an example of genetics shaping epigenetics. Reduced representation methods have demonstrated genome-wide differences in DNA methylation in multiple DS tissues and noted a group of genes with pan-tissue hypermethylation, which includes the clustered protocadherins and RUNX1. In our research, we were the first to apply low-pass WGBS to identify DMRs in DS brain samples and neonatal dried blood spots. By assaying regions of the genome that have never been examined before in DS patients, we demonstrated that many of the previously identified differentially methylated single CpG sites are actually part of larger scale DMRs with functional relevance to neurodevelopment. Machine learning feature selection refined these DMRs to 22 loci. We also observed differences related to the one-carbon metabolic pathway that establishes DNA methylation. Finally, we further characterized a dichotomy in the DNA methylation signature of DS, where many of the hypermethylated regions are observed in all tissues (pan-tissue) and many of the hypomethylated regions are tissue-specific. In order to functionally investigate the mechanism of the DS methylation signature, we have also established human neuronal cell culture models of DS (SH-SY5Y and LUHMES), which we transfected with the catalytically inactive but stimulatory DNA methyltransferase DNMT3L that is located on chromosome 21. We found that stable overexpression of DNMT3L recreates a significant proportion of the genome-wide pan-tissue DS DNA hypermethylation profile. However, DNMT3L overexpression alone is not sufficient to recreate the complete DNA methylation profile of DS. Notably, we also a observed a role for the known chromosome 21 encoded transcription factor RUNX1 in neonatal blood, where it was the most differentially methylated region in DS newborns and marked by a large block of hypermethylation that overlaps with its super-enhancer. We observed a genome-wide impact, as all of RUNX1’s binding sites were hypomethylated, which is in line with its known role in demethylating DNA at target genes. Overall, these experiments suggest that DNMT3L drives the pan-tissue hypermethylation profile in all DS tissues, while RUNX1 drives a tissue-specific hypomethylation profile. Although RUNX1 is primarily studied as a hematopoietic transcription factor, we also observed that it was hypermethylated in DS post-mortem brain, which is notable given its known but relatively underexplored role in neurodevelopment.